- Formula : C3IF2
-
Space Group : P2_1/c (14)
Centrosymmetric : True
Dimensionality : 1D -
Structure parametersa = 6.2187
b = 11.6168
c = 5.7876α = 90.0
β = 93.61
γ = 90.0 -
Number of atoms per primitive cell = 24
Total number of electrons per primitive cell = 132 -
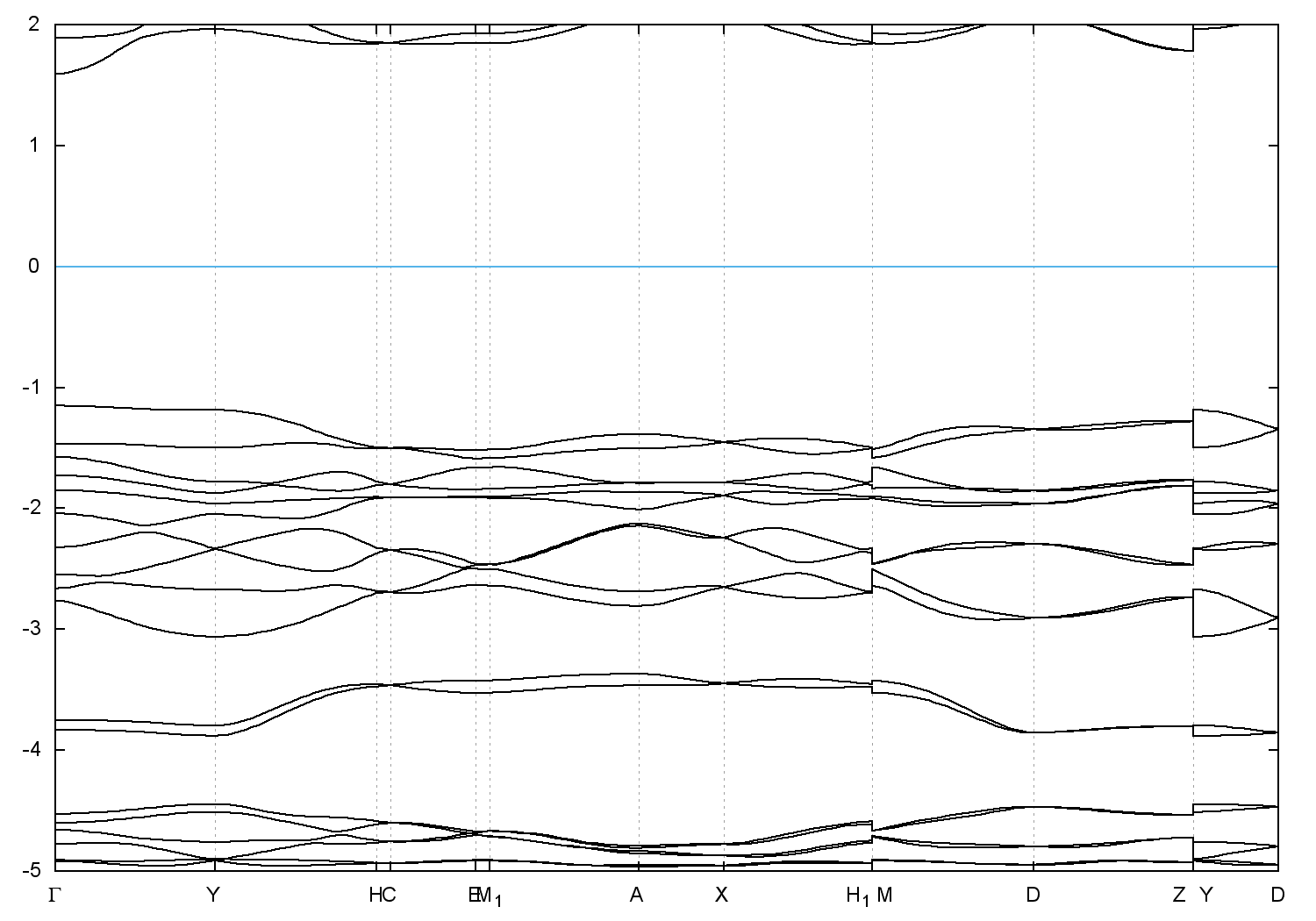
Band gap = 2.7461 eV
Direct Gap = 2.746 eV
Metallicity = 0.000
Topological Z2 indices ν = (0;000) - cif file - scf.in - scf.out - bands.in - bands.out
- Reference:
Switching between halogen- and hydrogen-bonding in stoichiometric variations of a cocrystal of a phosphine oxide,
CrystEngComm 14, 6110 (2012)
Band structure with spin-orbit coupling